Research
Our research applies primarily theoretical and computational approaches to understand the role that genomes play in macroevolutionary patterns and processes.
Inferring complex histories of speciation and introgression
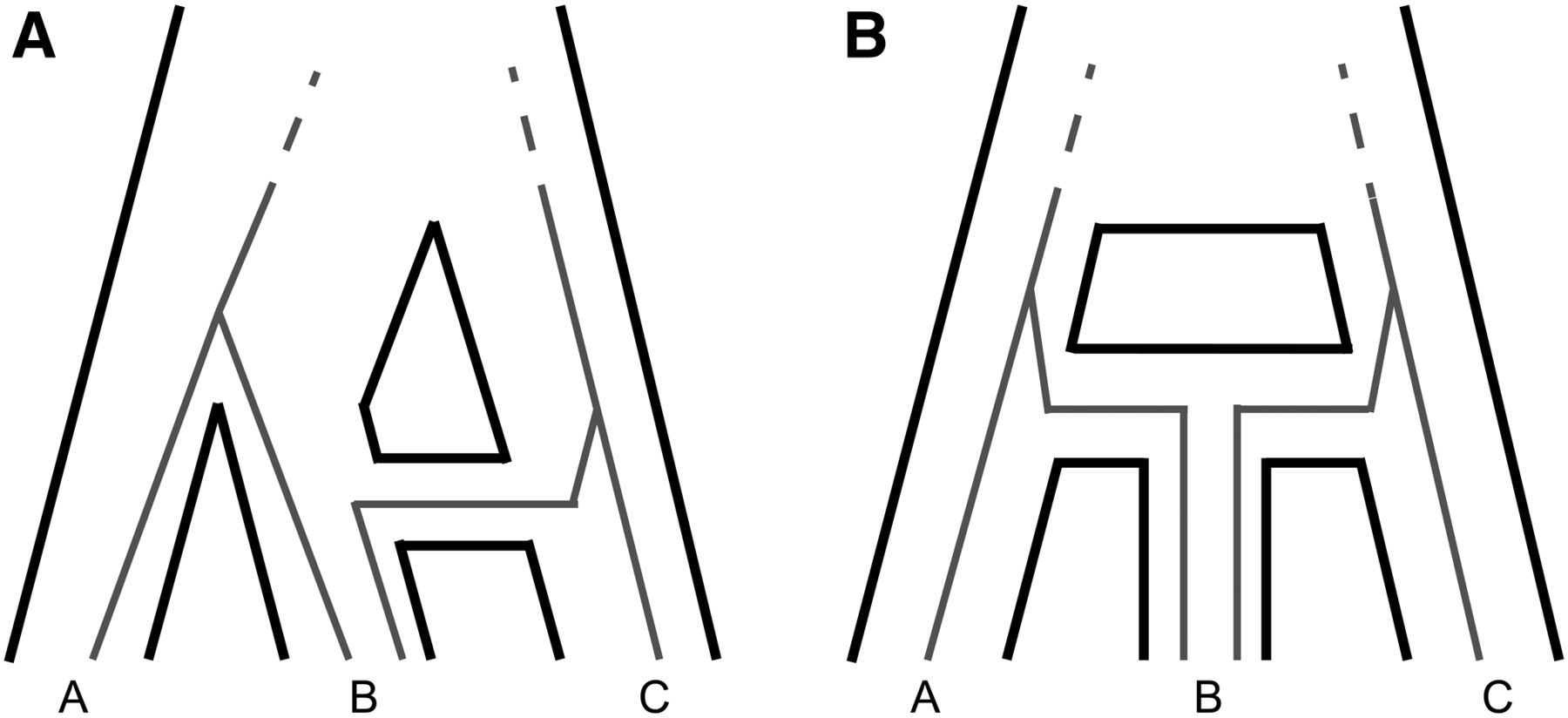
Modern genomic datasets across the tree of life have revealed a preponderance of hybridization and back-crossing among previously isolated lineages, a phenomenon called introgression. The high frequency of introgression is driving researchers to rethink the nature of species boundaries and our models of evolutionary relationships. Central to this endeavor is the creation of methods that can accurately detect and characterize introgression events. In my work, I develop mathematical models and statistical tools to make more detailed and biologically informed inferences about the occurrence, direction, timing, and biological factors affecting introgression. I am also interested in new approaches to species tree inference in the presence of introgression.
Phylogenetic comparative methods for discordant evolutionary histories
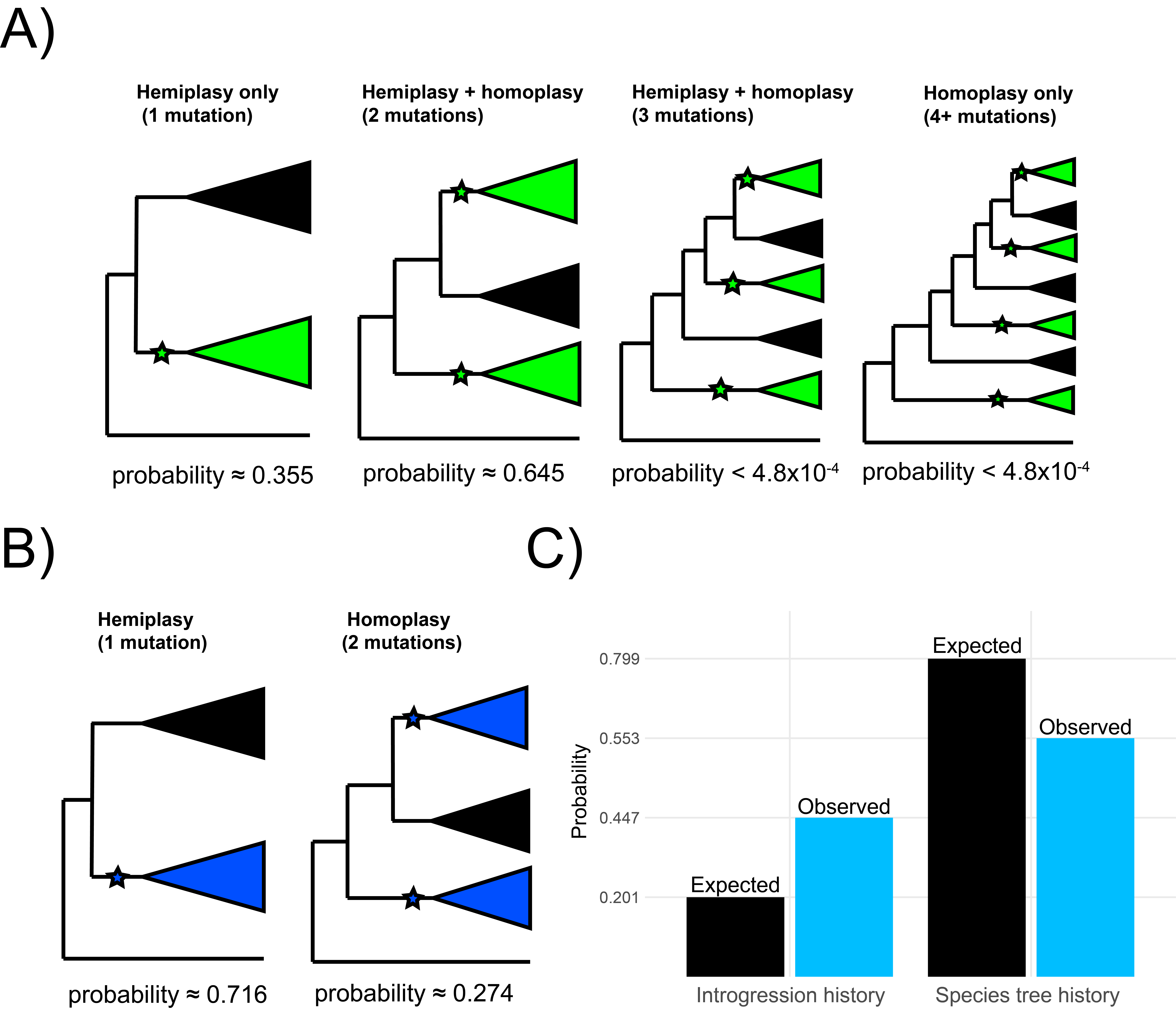
The statistical toolkit of phylogenetic comparative methods is essential to our understanding of processes of phenotypic evolution that play out over long timescales. These methods typically assume that a single, fixed species phylogeny can describe evolutionary relationships, an assumption that is increasingly being challenged by high rates of non-treelike patterns of relatedness such as incomplete lineage sorting and introgression. This can result in misleading comparative inferences if such patterns are not corrected for. I am interested in developing theory and software with the goal of making more robust comparative inferences in the presence of these complex patterns of common ancestry.
Understanding macroevolutionary drivers of genome and karyotype evolution
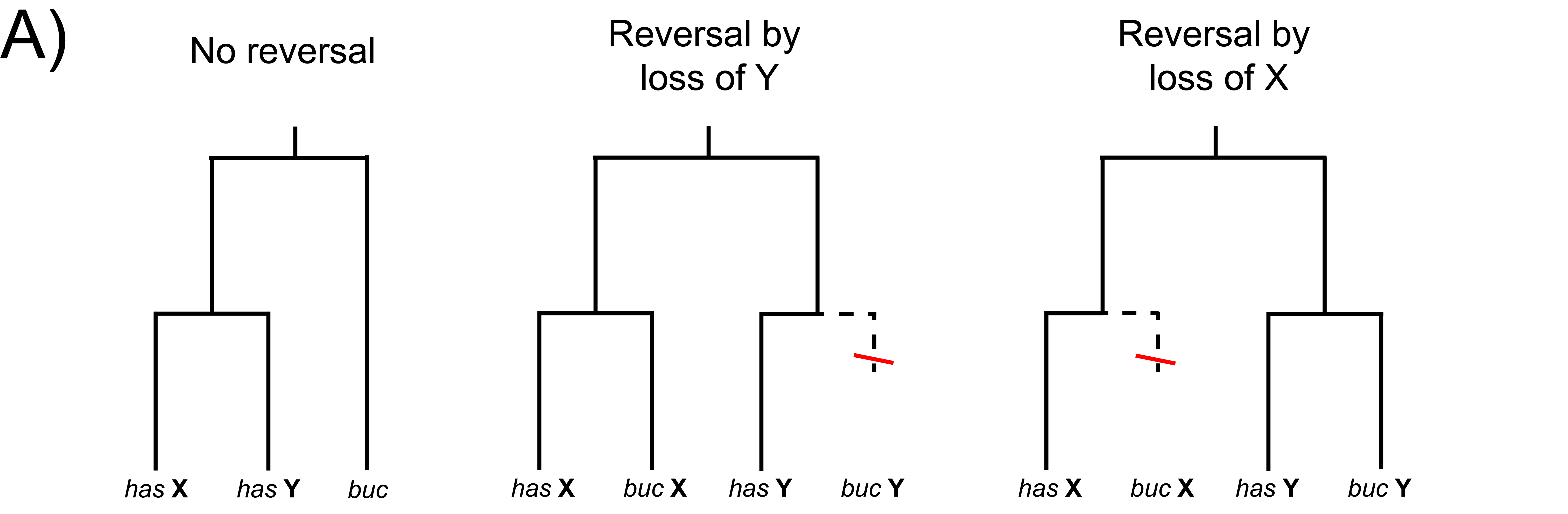
The structure and content of genomes varies widely among species. The forces shaping this variation are complex and, in many cases, not well understood. I am investigating these questions using a combination of theory, methods development, and modern genomic techniques in a variety of systems. A major focus of this work to date has been developing new comparative approaches to studying chromosomal rearrangements and the role they play in macroevolutionary processes like convergent evolution and speciation. I am also interested in investigating the drivers of sex chromosome gain, loss, and turnover, the rate of which varies widely across the eukaryotic tree of life.
The molecular basis of phenotypic convergence in complex quantitative traits
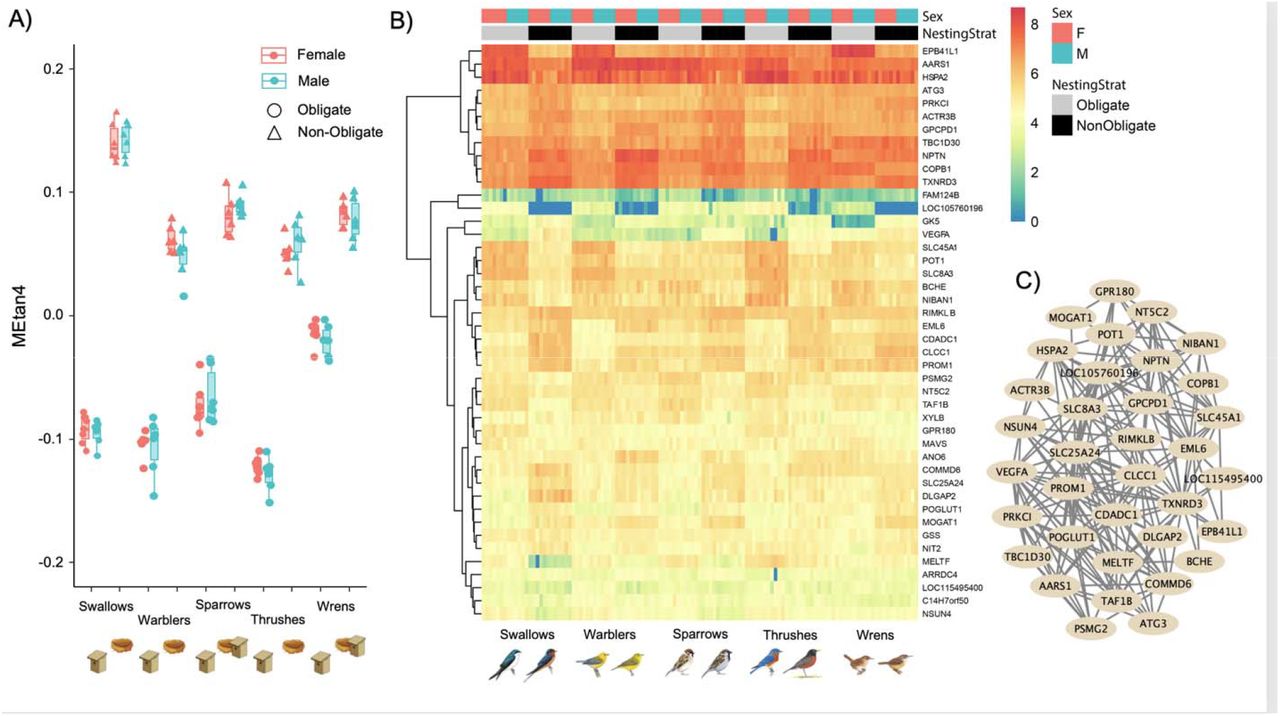
Advances in sequencing technology and computational resources are enabling the integration of phylogenetic comparative methodds with large-scale “omics” datasets. This is opening new avenues for investigations of the molecular genetic basis of convergent evolution in complex quantitative traits with highly polygenic genetic underpinnings. I am particularly interested in the evolution of gene expression across the whole transcriptome in response to such instances of phenotypic convergence. This work has been conducted primarily in collaborative efforts, with a current focus on the evolution of cavity nesting and territorial aggression in songbirds.